SpaGFT Tutorial:Visium Mouse Brain Coronal¶
Outline¶
Import packages
Load data
QC and preprocessing
Function: identify spatially variable genes
Function: gene expression enhancement
Function: characterize functional tissue units
The installation steps can be found here. SpaGFT is a python package to analyze spatial transcriptomics data via graph Fourier transform. To install SpaGFT, the python version is required to be >= 3.7. You can check your python version by:
[1]:
import platform
platform.python_version()
[1]:
'3.8.18'
1. Import packages¶
[2]:
import SpaGFT as spg
import numpy as np
import pandas as pd
import scanpy as sc
import matplotlib.pyplot as plt
import os
sc.settings.verbosity = 1
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor='white')
scanpy==1.9.1 anndata==0.9.2 umap==0.5.5 numpy==1.21.5 scipy==1.8.0 pandas==1.4.2 scikit-learn==1.0.2 statsmodels==0.14.1 python-igraph==0.9.10 louvain==0.7.1 pynndescent==0.5.11
Define the folder to save the results which are generated from the following analysis:
[3]:
results_folder = './results/mouse_brain_coronal_analysis/'
if not os.path.exists(results_folder):
os.makedirs(results_folder)
2. Load data¶
In this tutorial, we use a Visium dataset of Mouse Brain. The dataset can be downloaded here. Two key elements are essential for running SpaGFT: the raw gene counts matrix and spatial coordinates of spots. Indeed, the basic object input to SpaGFT should be an anndata object which contains the above two elements.
[5]:
adata = sc.read_visium("./data/mouse-he-coronal")
adata.var_names_make_unique()
# Add raw to anndata object
adata.raw = adata
You can also create your datasets which are anndata objects. Note that the raw count matrix should be found in adata.X, and the spatial coordinates of all spots should be found in adata.obs or adata.obsm. You can use diverse functions provided by scanpy to create the anndata object. For example, you can read Visium datasets by
sc.read_visium() # recommend
Alternatively, users can also create the anndata object using the raw count matrix and spatial coordinates of spots in a direct way:
count_mtx = pd.read_csv(PATH_TO_COUNT_MATRIX)
coord_mtx = pd.read_csv(PATH_TO_COORDINATE_MATRIX)
count_mtx.iloc[:5, :5]
coord_mtx.iloc[:5, :5]
# create the anndata object
adata = sc.AnnData(count_mtx)
adata.obs.loc[:, ['x', 'y']] = coord_mtx
adata.var_names_make_unique()
adata.raw = adata
More approaches to load datasets can be found at Scanpy
3. QC and preprocessing¶
Before the formal analysis, we perform some basic filtering of genes and normalize Visium counts data with the normalize_total method from Scanpy followed by log-transform.
[6]:
# QC
sc.pp.filter_genes(adata, min_cells=10)
# Normalization
sc.pp.normalize_total(adata, inplace=True)
sc.pp.log1p(adata)
4. Function: identify spatially variable genes¶
4.1 Determine the Fourier modes and identify SVGs¶
To begin with, determine the number of Fourier modes for detecting SVGs. SpaGFT provides the dermine_frequency_ratio function based on the kneedle algorithm to determine how many FMs used automatically. Note that ratio_low \(\sqrt{n}\)* and ratio_high \(\sqrt{n}\)* are the recommended low-frequency and high-frequency FMs, where :math:`sqrt{n}` is the number of spots in the original dataset.
[7]:
# determine the number of low-frequency FMs and high-frequency FMs
(ratio_low, ratio_high) = spg.gft.determine_frequency_ratio(adata,
ratio_neighbors=1)
Obatain the Laplacian matrix
In the following, SpaGFT will transform gene expression signals from the spatial domain to the frequency domain and analyze the frequency signals to identify SVGs.
[9]:
# calculation
gene_df = spg.detect_svg(adata,
spatial_info=['array_row', 'array_col'],
ratio_low_freq=ratio_low,
ratio_high_freq=ratio_high,
ratio_neighbors=1,
filter_peaks=True,
S=6)
# S determines the sensitivity of kneedle algorithm
# extract spaitally variable genes
svg_list = gene_df[gene_df.cutoff_gft_score][gene_df.fdr < 0.05].index.tolist()
print("The number of SVGs: ", len(svg_list))
# the top 20 SVGs
print(svg_list[:20])
The precalculated low-frequency FMs are USED
The precalculated high-frequency FMs are USED
Graph Fourier Transform finished!
svg ranking could be found in adata.var['svg_rank']
The spatially variable genes judged by gft_score could be found
in adata.var['cutoff_gft_score']
Gene signals in frequency domain when detect svgs could be found
in adata.varm['freq_domain_svg']
The number of SVGs: 2118
['Agt', 'Sparc', 'Epop', 'Nrgn', 'Mef2c', 'Camk2n1', 'Stx1a', 'Slc17a7', 'Kcnh3', 'Ngef', 'Igsf1', 'Dlk1', 'Ddn', 'Tcf7l2', 'Slc6a11', 'Arhgap36', 'Zfhx3', 'Itpka', 'Arpp19', 'Gpx3']
Detailly, spatially variable genes (SVGs) are genes that exhibit specific spatial patterns. SpaGFT will transform the gene expression signal from the spatial/vertex domain to the frequency/spectral domain and identify SVGs in the frequency domain. There are three key parameters: + ratio_neighbors, which determines the neighbors of a spot when construct the KNN graph. In SpaGFT, the top ratio_neighbors \(\sqrt{n}\)/2 nearest neighobors are used, where \(n\) represents the number of spots. + ratio_low_freq, which determines the number of low-frequency Fourier modes (FMs) used to describe the gene expression signals. In SpaGFT, the top ratio_low_freq * \(\sqrt{n}\) low-frequency Fourier modes will be utilized to describe gene expression signals, where \(n\) represents the number of spots.
ratio_high_freq, which determines the number of high-frequency Fourier modes used, as the contrast to measure how high the contributions of low-frequency to recover the signals of genes. In SpaGFT, the top ratio_high_freq * \(\sqrt{n}\) high-frequency Fourier modes will be utilized, where \(n\) represents the number of spots.
4.2 Visualize the identified SVGs¶
We will visualize several detected genes which exhibit diverse spatial patterns. This step can be achieved by sc.pl.spatial function provided by scanpy or our built-in scatter_gene function to visualize gene expression patterns.
[10]:
plot_svgs = ['Ahi1', 'Nsmf', 'Tbr1', 'Ano2', 'Cartpt', 'Cbln2', 'Ttr', 'Pmch']
sc.pl.spatial(adata, color=plot_svgs, size=1.6, cmap='magma', use_raw=False)

4.3 Plot frequency signals¶
One of the characteristics of SpaGFT is that it can obtain new representations for genes in the frequency domain. In above genes’ frequency signals can be found in adata.varm['freq_domain_svg'] and visualized by
[11]:
# use gene_freq_signal function to plot frequency signals
spg.plot.gene_freq_signal(adata, gene=plot_svgs[0])
spg.plot.gene_freq_signal(adata, gene=plot_svgs[4])


where the red signals correspond to low-frequency signals, while the blue signals correspond to high-frequency signals.
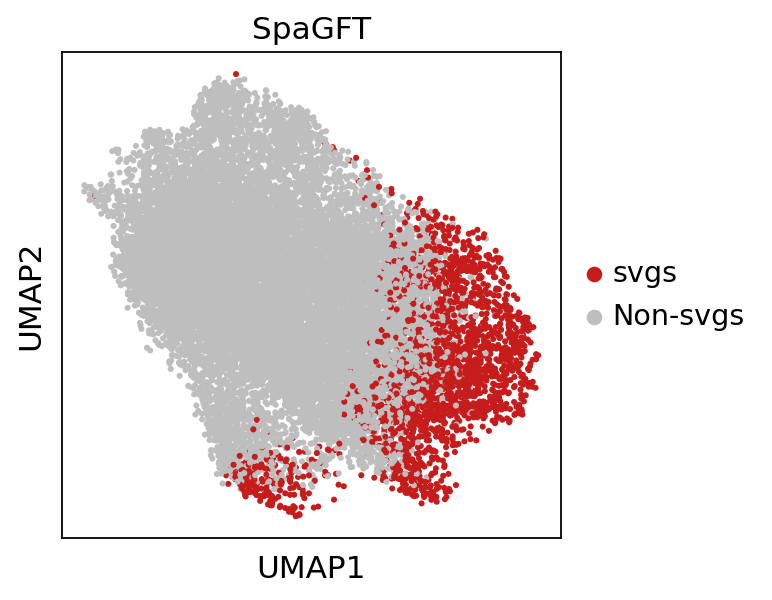
4.4 UMAP visualization of gene frequency signals¶
In addition, we can also separate SVGs and non-SVGs in the frequency domain. By projecting the frequency domain to 2-dimensional space by UMAP, we can verify this easily:
[12]:
# gene umap
spg.plot.gene_signal_umap(adata, svg_list, size=30)
svg_umap_df = pd.DataFrame(adata.varm['freq_umap_svg'],
index=adata.var_names,
columns=['UMAP_1', 'UMAP_2'])
The umap coordinates of genes when identify svgs could be found in
adata.varm['freq_umap_svg']
5 Function: gene expression enhancement¶
SRT expression data suffers from high noise, which will influence the credibility of analytical results. Therefore, SpaGFT adapts a low-pass filter to process signals in the frequency domain and performs inverse GFT to obtain enhanced signals. Note that this step is time-consuming and will take several minutes for Visium datasets.
[13]:
# copy from the original dataset
new_adata = adata.copy()
new_adata = spg.low_pass_enhancement(new_adata,
inplace=True,
c=0.01,
ratio_low_freq=15)
[14]:
# Before enhancement
sc.pl.spatial(adata, color=plot_svgs[:4], img_key=None, size=1.6, cmap='magma', use_raw=False)

[15]:
# After enhancement
sc.pl.spatial(new_adata, color=plot_svgs[:4], img_key=None, size=1.6, cmap='magma', use_raw=False)

In the following, we will use the enhanced gene expression matrix for analysis.
6. Function: identify functional tissue unit¶
6.1 Group SVGs based on frequency signals¶
The core of SpaGFT is that it can utilize SVGs to characterize functional tissue units by groups of SVGs with coincident spatial patterns. To achieve this, SpaGFT will analyze and process the signal’s low-frequency parts. In SpaGFT, the Louvain algorithm is used to cluster frequency signals of genes whose parameter resolution will be selected based on the designed objective function. Users can input the range of resolution by giving (start, end, step).
[16]:
# identify functional tissue units (FTU)
gene_df, new_adata = spg.identify_ftu(new_adata,
svg_list=svg_list,
ratio_fms=ratio_low,
resolution=(0.7, 1.8, 0.1),
ratio_neighbors=2,
n_neighbors=15)
resolution: 0.700; score: 0.1240
resolution: 0.800; score: 0.1238
resolution: 0.900; score: 0.1308
resolution: 1.000; score: 0.1306
resolution: 1.100; score: 0.1461
resolution: 1.200; score: 0.1472
resolution: 1.300; score: 0.1219
resolution: 1.400; score: 0.1044
resolution: 1.500; score: 0.1167
resolution: 1.600; score: 0.1466
resolution: 1.700; score: 0.1443
The above step will identify gene groups based on their spatial patterns. and the results can be found at adata.var.['tissue_module']. The genes in a group support a common spatial pattern, which is called a functional tissue unit. Besides, the functional tissue unit information can be found at adata.obsm['ftu_binary'].
[17]:
# save results
gene_df.to_csv(os.path.join(results_folder,
'mouse_brain_coronal_gene_information_results.csv'))
# the top 5 genes information
gene_df.iloc[:5, :]
[17]:
| gene_ids | feature_types | genome | n_cells | gft_score | svg_rank | cutoff_gft_score | pvalue | fdr | ftu | |
|---|---|---|---|---|---|---|---|---|---|---|
| Agt | ENSMUSG00000031980 | Gene Expression | mm10 | 1870 | 9.214738 | 1 | True | 4.197192e-97 | 7.565251e-95 | 6 |
| Sparc | ENSMUSG00000018593 | Gene Expression | mm10 | 2659 | 8.935703 | 2 | True | 1.006565e-81 | 1.043397e-79 | 6 |
| Epop | ENSMUSG00000043439 | Gene Expression | mm10 | 1378 | 8.804805 | 3 | True | 4.673493e-45 | 1.823850e-43 | 2 |
| Nrgn | ENSMUSG00000053310 | Gene Expression | mm10 | 2645 | 8.708655 | 4 | True | 1.864018e-118 | 1.094928e-115 | 3 |
| Mef2c | ENSMUSG00000005583 | Gene Expression | mm10 | 2319 | 8.687262 | 5 | True | 9.824203e-105 | 2.493936e-102 | 2 |
The last column indicates the supported functional tissue units.
6.2 Visualize clustering results¶
We can visualize the clustering results by UMAP:
[18]:
spg.plot.scatter_umap_clustering(new_adata, svg_list)

There are twelve functional tissue units via our computation. In the following, we will visualize the results.
6.3 Extract functional tissue unit information¶
To begin with, obtain functional tissue unit information by
[19]:
# The functional tissue unit information can be obtained in
ftu_binary_df = new_adata.obsm['ftu_binary']
ftu_binary_df.to_csv(os.path.join(results_folder,
'Lymphnode_tissue_module_information_results.csv'))
ftu_binary_df.iloc[:5, :10]
[19]:
| ftu_1 | ftu_10 | ftu_11 | ftu_12 | ftu_2 | ftu_3 | ftu_4 | ftu_5 | ftu_6 | ftu_7 | |
|---|---|---|---|---|---|---|---|---|---|---|
| AAACAAGTATCTCCCA-1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 |
| AAACAATCTACTAGCA-1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| AAACACCAATAACTGC-1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| AAACAGAGCGACTCCT-1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 |
| AAACCGGGTAGGTACC-1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
The rows of the above dataframe indicate the spots, and the columns indicate the functional tissue unit. Among them, ‘1’ reflects the structure of a functional tissue unit.
6.4 Visualize interested FTUs¶
We can visualize the interested functional tissue units by built-in scatter_ftu function:
[20]:
# Plot FTU-5 FTU-6 FTU-12
spg.plot.scatter_ftu(new_adata,
ftu=['ftu_6', 'ftu_8', 'ftu_9'],
spatial_info='spatial',
size=15)

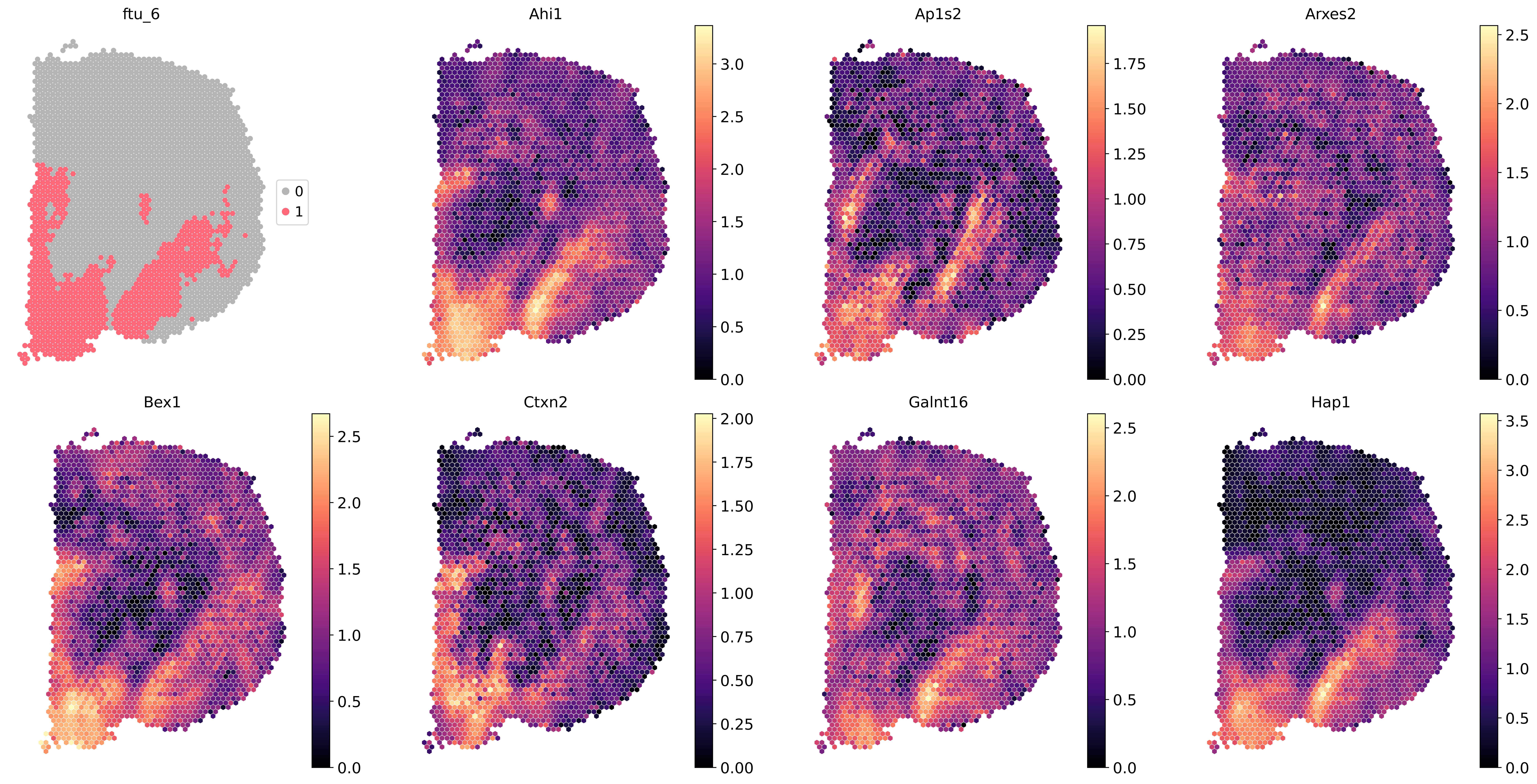
Precisely, we can also visualize functional tissue units and genes that support corresponding functional tissue units:
[21]:
ftu6_genes = gene_df.loc[gene_df.ftu=='6', :].index.tolist()
# plot FTU 6 and corresponding genes
# ['Ahi1', 'Ap1s2', 'Arxes2', 'Bex1', 'Ctxn2', 'Galnt16', 'Hap1']
plot_ftu6_genes = [ftu6_genes[19], ftu6_genes[74], ftu6_genes[156], ftu6_genes[15],
ftu6_genes[38], ftu6_genes[170], ftu6_genes[12]]
spg.plot.scatter_ftu_gene(new_adata,
ftu="ftu_6",
gene=plot_ftu6_genes,
spatial_info='spatial',
size=15)
6.5 Enrichment analysis of biological process¶
A group of genes that support a functional tissue unit has a similar spatial pattern, and we can perform functional enrichment analysis to explore the underlying biological process under this pattern.
[22]:
import gseapy as gp
ftu6_gene_list = gene_df.loc[gene_df.ftu == '6', :].index.tolist()
enr = gp.enrichr(gene_list=ftu6_gene_list,
gene_sets=['BioPlanet_2019','GO_Biological_Process_2021'],
organism='Mouse',
description='Tissue_module',
outdir='results/mouse_brain_coronal_analysis/enrichr_kegg',
no_plot=False,
cutoff=0.5 # use lower value from range(0,1),
)
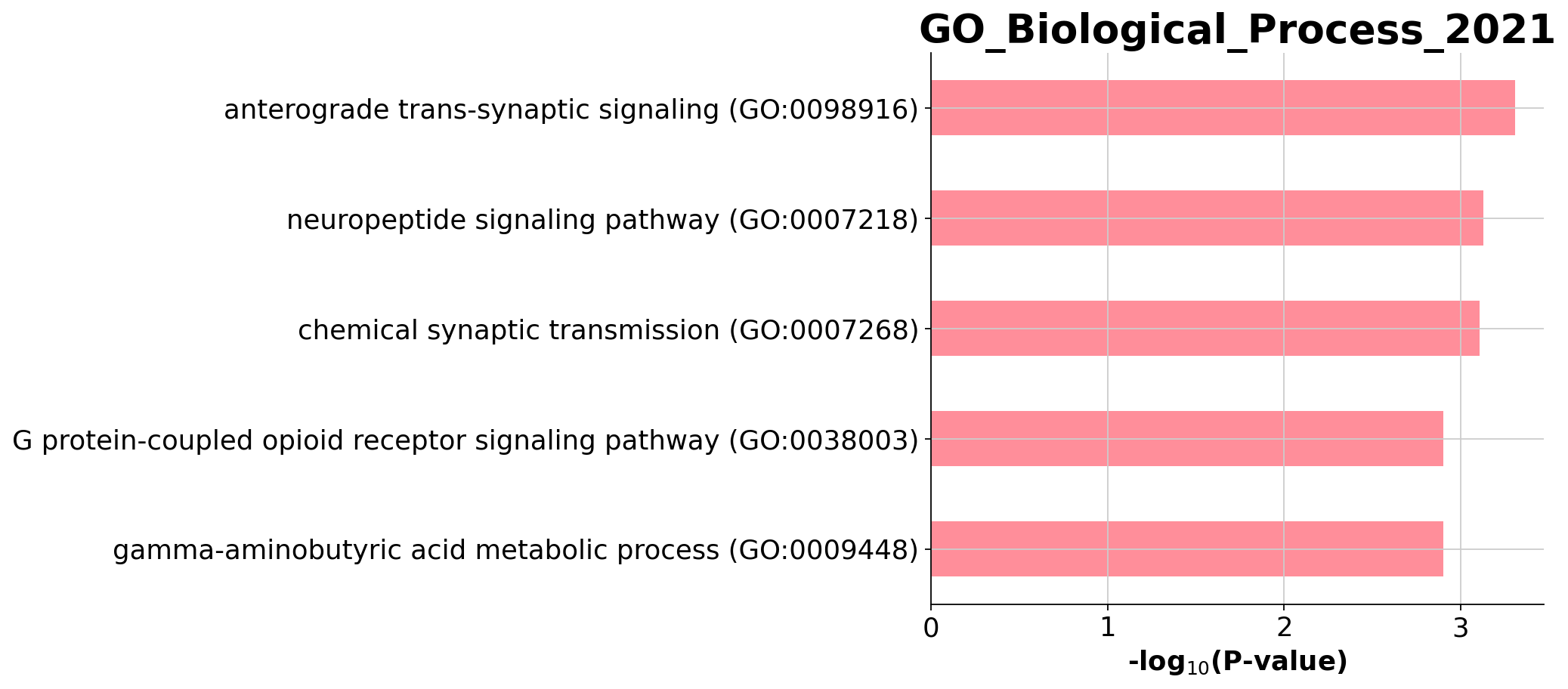
The enriched biological process of FTU 6:
[23]:
enr_results = enr.results
from gseapy.plot import barplot, dotplot
barplot(enr.results[enr.results.Gene_set=='GO_Biological_Process_2021'],
column='P-value',
color='#FF6879',
top_term=5,
title='GO_Biological_Process_2021')